CE認證是歐盟的產品安全認證,所有進入歐盟市場的醫療器械都必須進行醫療器械CE認證,醫療器械需要滿足的CE指令有《有源植入性醫療器械指令》(AIMDD, 90/385/EEC)、《醫療器械指令》(MDD,93/42/EEC)和《體外診斷器械指令》(IVDD, 98/79/EC)。
醫療器械CE認證包括哪四方面 ?
CE認證是一個完善的安全保障系統,并非僅僅是將一個樣品拿到試驗室檢驗通過而已。
因為 CE 標志是一個安全標志,所以,一個通過CE認證的產品必須確保自產品 的設計,生產,包裝,說明書的編寫,到運輸,銷售,產品的整個有效使用壽命 中,以及使用后產品的回收,等等所有環節中,均符合歐洲的健康、安全、與 環境保護之相關法律中所規定的基本要求。因此,一家制造商欲想使其產品通過 CE認證,通常要滿足如下4方面的要求:
1.產品投放到歐洲市場前,在產品上加貼CE標簽。
2.產品投放到歐洲市場后,技術文件(Technical Files)必須存放于歐盟境 內供監督機構隨時檢查。
3.對被市場監督機構發現的不合CE要求的產品、或者使用過程中出現事故但是已加貼CE標簽的產品,必須采取補救措施。(比如從貨架上暫時拿 掉,或從市場中永久地撤除)。
4.已加貼 CE標簽之產品型號在投放到歐洲市場后,若遇到歐盟有關的法律更改或變化,其后續生產的同型號產品也必須相應地加以更改或修正,以便符合歐盟新的法律要求。

CE 認證程序
1. 確認出口國家
2. 確認產品類別及歐盟相關產品指令
3. 指定“歐盟授權代表( 歐盟授權代理 ) ”(Authorized Representative)
4. 確認認證所需的模式(Module)
5. 采用 " 自我聲明 " 模式還是 " 必須通過第三方認證機構"
6. 建立技術文件 (Technical Files) 及其維護與更新
1、確認出口國家
若出口至歐洲經濟區EEA包括歐盟EU及歐洲自由貿易協議EFTA的 30 個成員國 中的任何一國,則可能需要CE認證。
2、確認產品類別及歐盟相關產品指令
若產品屬于這里所列22 類中的任何一個,一般地講,則需要進行 CE認證。若一個產品同時屬于一個以上的類別,則必須滿足所有類別相對應的產品指令中所列出的要求。
注: 某些產品指令中有時會列出一些排除在指令外的產品。
3、指定“歐盟授權代表( 歐盟授權代理 ) ”(Authorized Representative)
為了能確保前述CE標志 (CE Marking )認證實施過程中的4 項要求得以滿足,歐盟法律要求位于30 個 EEA 盟國境外的制造商 必須在歐盟境內指定一家歐盟授 權代表 ( 歐盟授權代理 ) (AuthorizedRepresentative),以確保產品投放到歐洲 市場后,在流通過程及使用期間產品“安全”的一貫性;技術文件 (Technical Files)必須存放于歐盟境內供監督機構隨時檢查;對被市場監督機構發現的不 合 CE要求的產品、或者使用過程中出現事故但是已加貼CE標簽的產品,必須 采取補救措施。(比如從貨架上暫時拿掉,或從市場中永久地撤除);已加貼 CE標簽之產品型號在投放到歐洲市場后,若遇到歐盟有關的法律更改或變化,其后續生產的同型號產品也必須相應地加以更改或修正, 以便符合歐盟新的法律 要求。
4、確認認證所需的模式(Module)
對于幾乎所有的歐盟產品指令來說,指令通常會給制造商提供出幾種 CE認證 (Conformity Assessment Procedures)的模式 (Module) ,制造商可根據本身的情況量體裁衣,選擇最適合自已的模式。一般地說,CE 認證模式可分為以下9種基本模式 :
1.Module A: internal production control
模式 A: 內部生產控制 ( 自我聲明 )
2.Module Aa: intervention of a Notified Body
模式 Aa: 內部生產控制 加第 3 方檢測
3.Module B: EC type-examination
模式 B: EC型式試驗
4.Module C: conformity to type
模式 C: 符合型式
5.Module D: production quality assurance
模式 D: 生產質量保證
6.Module E: product quality assurance
模式 E: 產品質量保證
7.Module F: product verification
模式 F: 產品驗證
8.Module G: unit verification
模式 G: 單元驗證
9.Module H: full quality assurance
模式 H: 全面質量保證
基于以上幾種基本模式的不同組合,又可能衍生出其它若干種不同的模式。一 般地說,并非任何一種模式均可適用于所有的產品。換言之, 也并非制造商可以隨意選取以上任何一種模式來對其產品進行CE認證。
5、采用“自我聲明”模式還是“必須通過第三方認證機構”
風險水平 (Risk Level) 較低 (Minimal Risk)
歐盟的產品指令允許某些類別中風險水平 (Risk Level) 較低 (Minimal Risk) 的產品之制造商選擇以模式A:“內部生產控制 ( 自我聲明 ) ”的方式進行CE 認證。
風險水平較高的產品必須通過第三方認證機構 NB(Notified Body) 介入。
對于風險水平較高的產品,其制造商必須選擇模式 A以外的其它模式, 或者模式A外加其它模式來達到 CE 認證。也就是說,必須通過第三方認證機構NB(Notified Body)介入。
模式 A 以外的其它模式的認證過程中,通常均需要至少一家歐盟認可的認證機構 NB 參于認證過程中的一部分或全部。根據不同的模式,NB則可能分別以:來樣 檢測,抽樣檢測,工廠審查,年檢,不同的質量體系審核,等等方式介入認證過 程,并出具相應的 檢測報告,證書等。
目前,已經有 1200 多家認證機構獲得歐盟認可, 這些認證機構中的絕大多數位 于歐盟盟國境內。通常情況下, 一家 NB僅被歐盟授權可針對某一類或幾類產品 進行某一或幾種模式下的認證。換言之,一家歐盟授權的認證機構并不可能針 對所有的產品種類進行認證,即使對其被授權的產品種類,通常情況下也并非 被授權所有的模式。對于每一個歐盟的產品指令,通常都有一個針對該產品指 令的授權認證機構NB名錄。
6、建立技術文件 (Technical Files)及其維護與更新
歐盟法律要求,加貼了 CE標簽的產品投放到歐洲市場后, 其技術文件 (TechnicalFiles) 必須存放于歐盟境內供監督機構隨時檢查。技術文件中所包涵的內容若有變化,技術文件也應及時地更新。
"技術文檔"是歐盟醫療器械指令中很重要的一個事項, 它的目的是要求企 業準備充份的技術資料和證明, 供主管機關抽查, 或發生訴訟糾紛時使用。 各歐盟指令對于 " 技術檔案 " 的要求有所差別, 在這里謹以中國出口企業最 常用的“醫療器械”的要求為例,加以說明。
醫療器械指令 93/42/ EEC要求 " 技術檔案 " 可能包含下列項目:
A、企業的質量手冊和程序文件
B、企業簡介及歐洲代理名稱、聯系方式
C、CE符合性聲明(或稱自我保證聲明, 若該產品是和其它設備聯合運用, 則應有整體符合基本要求的證明材料)
1.產品名稱、分類及引用標準條款的簡要描述
2.產品概述(包括類型和預期用途)
a) 產品的歷史沿革
b) 技術性能參數
c) 產品配合使用的附件、配合件和其它設備清單
d) 產品的圖示與樣品
e) 產品所用原材料及供應商
3.使用該產品的調和標準 / 或其它標準
4.風險分析評估結論和預防措施( EN1441 產品服務危險分析報告)
5.生產質量控制
a) 產品資料和控制文檔(包括產品生產工藝流程圖)
b) 產品的滅菌方法和確認的描述
c) 滅菌驗證
d) 產品質量控制措施
e) 產品穩定性和效期的描述
6.包裝和標識
a) 包裝材料說明
b) 標簽
c) 使用說明書
7.技術評價
a) 產品檢驗報告及相關文獻
b) 技術概要及權威觀點
8.潛在風險評價
a) 產品潛在風險測試報告及相關文獻
b) 潛在風險的概要及權威觀點
9.臨床評價
a) 產品臨床測試報告及相關文獻
b) 臨床使用概述及權威觀點
附錄 1、產品出廠檢測報告
附錄 2、產品型式檢測報告
附錄 3、基本要求檢查表
注:
1、臨床研究(包括:物理性能,生化、藥理 、藥動及毒性研究,功 效測試,滅菌合格證明,藥物相容性等)
2、生物兼容性測試
( A)EN30993 第一部分要求:細胞毒性、感光性、刺激 - 皮內反應、急性 全身中毒、致熱性、亞急性中毒、遺傳毒性、植入溶血性;
( B)支持測試:慢性中毒、致癌性、再生性 / 生長性毒素、生物動因退化。)
3、臨床資料(需要臨床研究或描述臨床研究)
4、包裝合格證明(EN868)
5、標簽、使用說明(EN980、EN1041)
6、結論(設計檔案資料的接受、利益對應風險的陳述)
上述文件都必須用歐盟官方語言之一 ( 英、德、法文 ) 編寫,但使用說明必須用使用者所在國語言編寫。所有文件應在最后一次出貨后,至少保存五年。
體外診斷醫療器械 IVDD產品分類
根據 98/79/EC( IVDD)指令附錄2 確定產品分類原則對有認證需求的產品進行分類。分類的依據是產品所診斷的疾病。常見產品的分類可參考下表:

與上述診斷試劑配套使用的校準品、儀器、標本采集保存用具均屬于體外診斷器 械指令管理的范疇。
醫療器械 MDD產品分類
CE認證過程中判斷一個醫療器械正確的分類,僅憑器械的名稱是不夠的,必須知道完整的預期使用目的(Intended Purpose)!
我們經常聽到這樣的一句話問題: 某某產品在 CE分類里屬于幾類醫療器械? 提問者也許不知道僅從一個醫療器械的名稱而判斷其 CE認證過程中的分類經 常是不妥當的!
1、歐盟與美國的區別 歐盟與美國的醫療器械的分類有很大的不同。
美國的 FDA將醫療器械根據其通用的特點 事先已經分類 并建立了一個公開的數據庫可查詢;
歐盟則是建立了一套分類規則,讓制造商根據產品的預期使用目的(Intended Purpose)按照分類規則自己進行分類。
2、同一個產品,既可以是醫療器械,也可以不是醫療器械
在美國,一個產品是否為醫療器械完全由 FDA決定;
在歐盟,一個產品是否為醫療器械由制造商 ( 申明的產品預期使用目的) 決定 , 比如:電熱褥既可以是醫療器械,也可以不是醫療器械。
3、同一個產品,可以是不同類別的醫療器械
比如: 制造商申明的預期使用目的不同, 電熱褥既可以是 I 類醫療器械, 也可以是 IIa 或 IIb類醫療器械。
4、同一個產品,作為系統的一部分時與作為配件時屬于不同的類別
比如:手術過程中用非主動式抽取腹水裝置的留在體外的盛腹水的容器, 作為系統的一部分時可屬于IIa 類,但是作為配件時則可屬于I 類。
5、類似的產品,可以是不同類別的醫療器械
比如: X光拍片時常用的圖像儲存通信系統Picture Archiving and Communication Systems (PACS) ,不同制造商申明的預期使用目 ( 功能) 的不同, PACS可以是 I 類醫療器械,也可以是IIa 或 IIb 類醫療器械。
6、類似的產品,有的屬于醫療器械 MD, 有的則屬于體外診斷器械 IVD
比如:采血管如果 是侵入式的或接觸到皮膚的,則屬于 MDD 93/42/EEC指令管轄的(普通)醫療器械 MD;
如果 是非侵入式的或完全接觸不到皮膚的,則屬于IVD 98/79/ec 指令管轄的體外診斷器械IVD。
醫療器械指令 MDD 93/42/eec附錄九中詳定18 條規則,按醫療產品的危險程度,將產品分為Ⅰ類、Ⅱa類、Ⅱb類、Ⅲ類。
產品分類規則:
1、規則應用由器械的預期使用目的決定;
2、如果器械是和其它器械配合使用,分類規則分別適用于每種器械;
3、附件可以和其它一起使用的器械分開單獨分類;
4、啟動或影響某種器械的軟件與器械屬于同一類型。
分類準則:
時間:暫時(<60 分鐘)、短期(<30 天)、長期( >30 天)
創傷性:非創傷、通過孔徑創傷,外科創傷 、植入。
適用位置:中央循環、中樞神經系統,其它地方。
能量供應:無源,有源。
規則 1~4、所有非創傷性器械均屬于I 類,除非他們:
用于儲存體液 ( 血袋例外) II a 類
于 Ila類或更高類型的有源醫療器械類II a 類
改變體液成分 II a / II b 類
一些傷口敷料 II a / II b 類
規則 5、侵入人體孔徑的醫療器械
暫時使用(牙科壓縮材料、檢查手套 ) I類
短期使用(導管、隱形眼鏡)II a類長期使用(正常牙線) II b類
規則 6-8 、外科創傷性器械
再使用的外科器械(鉗子,斧子) I類
暫時或短期使用(縫合針。外科手套) 11a類
長期使用(假關節,眼內晶體 )II b 類
與中央循環系統 (CCS)或中樞神經系統接觸的器械 III 類
規則 9、給予或交換能量的治療器械 II a 類
(肌肉刺激器、電鉆、皮膚光療機、助聽器)
一種潛在危險方式工作的II b 類
(嬰兒培養箱、高頻電刀、超聲碎石機、 X 光機)
規則 10、診斷器械
提供能量( 核磁共振,超聲診斷儀)IIa 類
診斷/監視體內放射藥物分布 II a 類
(r照相機、正電子發射成像儀)
診斷/監視生理功能(心電圖、腦電圖) II a 類
危險情況下監視生理功能II b 類
(手術中的血氣分析儀)
發出電離輻射 (X 射線診斷議) II b 類
規則 11 控制藥物或其他物質進出人體的有源器械 II a 類
(吸引設備、供給泵)
如以一種潛在危險方式工作 II b 類
(麻醉機、呼吸機、透析機、高壓氧艙)
規則 12 所有其他有源醫療器械屬于I 類
(觀察燈、牙科椅、輪椅、牙科用治療燈、記錄處理觀察診斷圖象用的 有源器械)
規則 13、與醫用物質結合的器械(含殺精子的避孕套、含抗生素的牙髓材料)III類
規則 14、避孕用具(避孕套、子宮帽II b 類 ) II b/III 類 (子宮內避孕器III 類)
規則 15、清洗或消毒的器械
醫療器械 ( 內窺鏡消毒 ) II a 類
接觸鏡 ( 消毒液、護理液 ) II a 類
規則 16、用于記錄X射線圖象的器械 (X 光片 ) II a 類
規則 17、利用動物組織的器械(生物)心臟瓣膜、腸線、膠原) III 類
規則 18、血袋 II b 類
各種類型醫療器械的 CE認證步驟
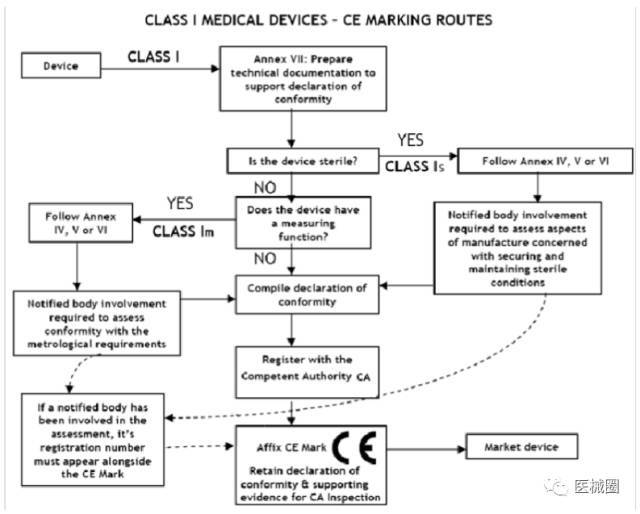
I 類醫療器械的 CE認證步驟
1、分類:確認產品屬于I 類醫療器械
2、選擇符合性評估途徑:請參考下面的流程圖
3、編制技術文件
4、CE符合性聲明
5、委任歐盟授權代表
6、由歐盟授權代表將制造商及產品在歐盟主管機關注冊
7、建立售后警戒系統/ 加貼 CE標簽并將產品投放市場
I類醫療器械: CE 符合性評估途徑
1、制造商有責任確保其產品符合93/42/eec指令的所有相關的基本要求,必須制定一 份書面(自我)聲明來保證。
2、不具備測量功能或非滅菌的 I 類醫療器械(的 CE認證過程中)不需要第三方公告機 構(NB) 參與。 是否符合ISO13485:2003標準,由制造商自愿選擇,并非強制性。
3、具有測量功能或滅菌類的 I 類醫療器械(的 CE認證過程中)必須要有第三方公告機 構(NB) 參與。
4、一旦制造商認為其產品符合 93/42/eec 指令的所有相關的基本要求,(歐盟境內的) 制造商,或者(歐盟境外制造商的)歐盟授權代表必須先在歐盟主管機關注冊, 然 后才可 加貼 CE標簽并將產品投放EEA市場。
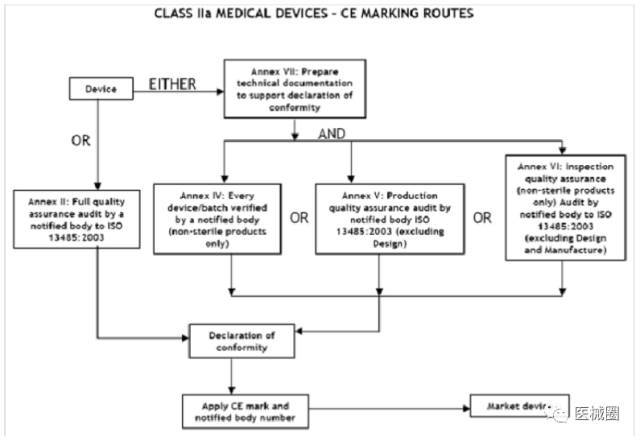
IIa類醫療器械的CE認證步驟
1、分類:確認產品屬于IIa類醫療器械
2、選擇符合性評估途徑:請參考下面的流程圖
3、編制技術文件
4、委任歐盟授權代表
5、從第三方公告機構(NB) 獲得 CE證書
6、( 完成 )CE 符合性聲明
7、將技術文件存放在歐盟授權代表處( 供歐盟主管機關隨時檢查 )
8、建立 ( 售后 ) 警戒系統/ 加貼CE標簽并將產品投放 EEA市場
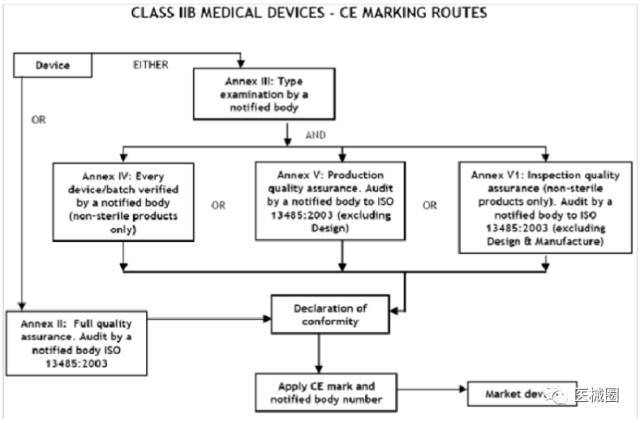
IIb類醫療器械的CE認證步驟
1、分類:確認產品屬于IIb 類醫療器械
2、選擇符合性評估途徑:請參考下面的流程圖
3、編制技術文件
4、委任歐盟授權代表
5、從第三方公告機構(NB) 獲得 CE證書
6、( 完成 )CE 符合性聲明
7、將技術文件存放在歐盟授權代表處( 供歐盟主管機關隨時檢查 )
8、建立 ( 售后 ) 警戒系統 / 加貼CE標簽并將產品投放EEA市場
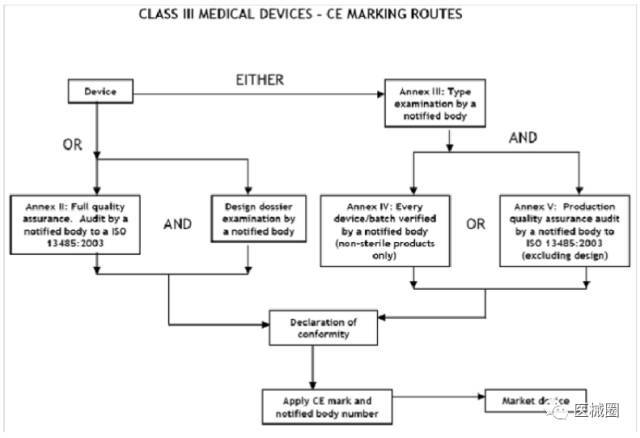
III類醫療器械的CE認證步驟
1、分類:確認產品屬于III類醫療器械
2、選擇符合性評估途徑:請參考下面的流程圖
3、編制技術文件
4、委任歐盟授權代表
5、從第三方公告機構(NB) 獲得 CE證書
6、( 完成 )CE 符合性聲明
7、將技術文件存放在歐盟授權代表處( 供歐盟主管機關隨時檢查 )
8、建立 ( 售后 ) 警戒系統 / 加貼CE標簽并將產品投放EEA市場